Site Radar
Site Radar is a graph neural network-based module designed for identifying protein-ligand binding sites. It utilizes machine learning techniques to predict and classify potential binding pockets based on the protein structure.
During this stage, you can configure the following key parameters:
- Approach: Defines the type of neural network and features used for site prediction.
- Minimum Pocket Size: Determines the smallest allowed cluster of annotated grid points to be considered a potential binding site.
Approach Selection
-
AA-Specific Approach
- Utilizes both shape and amino acid composition of the target protein.
- Recommended to be preferred in case of binding sites with non-canonical shape, such as shallow solvent-exposed pockets.
-
Geometric Approach
- Uses only the shape of the protein to identify potential binding sites.
- Less selective, typically identifying more pockets than the AA-specific approach.
- Recommended for pockets with unusual amino acid distributions.
Minimum Pocket Size
Site Radar employs a dot-based approach, forming volumetric binding sites by clustering grid points annotated by the neural network.
- The minimum pocket size parameter determines the smallest cluster of points required for a binding site to be considered valid.
- The default value was selected based on extensive benchmarking and experimental validation.
- If no binding site is found with standard settings, decreasing this parameter may help detect smaller pockets.
Note: Site Radar does not generate binding pockets in regions occupied by cofactors. Ensure that cofactors are correctly annotated in the protein structure to avoid incorrect results.
For further details, refer to the publication:
Site Radar: Utilizing Graph Machine Learning for Precise Mapping of Protein–Ligand-Binding Sites
(DOI: 10.1021/acs.jcim.2c01413).
Pocket Selection
By default, all detected binding pockets for the target protein are displayed in semi-transparent colors, allowing easy navigation.
-
To select a pocket, use the colored sidebar and click the box corresponding to the pocket you wish to analyze.
-
The selected pocket will become opaque in the 3D visualization.
Pocket Extension
Once a pocket is selected, you can extend its size.
- Pocket size adjustment is performed using the slider.
- The selected number represents the expansion distance in angstroms (Å) in all directions.
- The extension considers potential clashes with the protein and cofactors (if present).
Pocket Characteristics
Pocket properties are displayed in a diagram and provide key insights into ligand-binding potential.
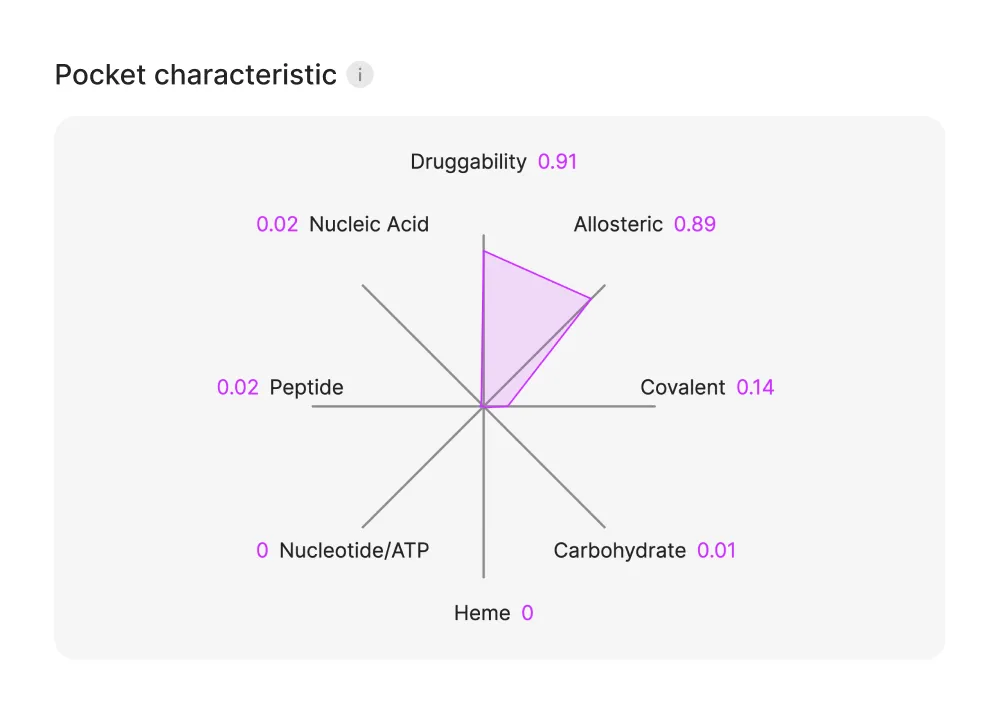
The characteristics include:
-
Druggability – Assesses whether the binding site is likely to accommodate a drug-like molecule (calculated by Site Radar).
-
Allosteric – Indicates the potential for binding allosteric modulators.
-
Covalent – Evaluates the ability to bind covalent ligands.
-
Carbohydrate – Determines the potential for carbohydrate binding.
-
Heme – Checks the ability to bind heme groups.
-
Nucleotide/ATP – Identifies affinity for nucleotides and ATP molecules.
-
Peptide – Detects if the site can bind peptides.
-
Nucleic Acid – Evaluates the potential for nucleic acid interactions.
Note:
- Pocket characteristics are only available for pockets generated by the Site Radar module.
- Extended pockets do not have calculated characteristics.
Merging Pockets
If multiple pockets are available, users can merge adjacent pockets by enabling merge mode.
Merging Process:
-
Enable merge mode to start merging.
-
Select two or more pockets that you want to merge (incompatible pockets will appear blurred).
-
The selected pockets will appear opaque in the 3D visualization.
-
Press the “Merge” button to combine the pockets.
Important Notes:
- If a pocket was extended before merging, its expansion will be reset after merging.
- Merged pockets are listed in the merge history section.

- The merging can be undone by pressing the “X” symbol next to the merged pocket.
- Extended merged pockets will not have calculated pocket characteristics.