Scaffold Generation and Periphery Generation
The Scaffold Generation and Periphery Generation modules are used in different stages of molecular structure design. Scaffold Generation is responsible for creating the molecular core based on selected probes, while Periphery Generation expands this core by attaching functional groups.
Scaffold Generation
This module constructs a molecular scaffold starting from selected probes within the binding site.
- Probes are selected as the starting points for scaffold generation.
- The resulting structure maintains pre-defined interactions within the binding pocket.
- The module ensures that the generated scaffold remains drug-like.
Users can customize the process by adjusting basic and advanced parameters.
Basic Parameters
1. Maximum Execution Time (1–168 h, Default: 96 h)
Sets the maximum runtime for scaffold generation. Longer times allow more extensive searching but increase computation time.
2. Maximum Heavy Atoms to Add (1–30, Default: 20)
Defines the maximum number of non-hydrogen atoms that can be introduced into the scaffold.
3. Types Limit
Controls the maximum number of each atom type allowed in the final scaffold.
| Atom Type | Example | Description |
|---|---|---|
| .=O | 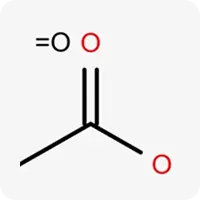 | Oxygen atom in sp² hybridization, acts as a hydrogen bond acceptor |
| C2r | 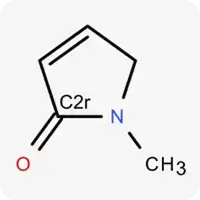 | Carbon atom in sp² hybridization within a non-aromatic ring |
| C3r | 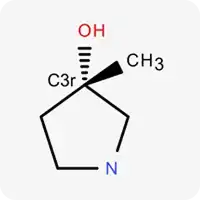 | Carbon atom in sp³ hybridization within a ring |
| Car | 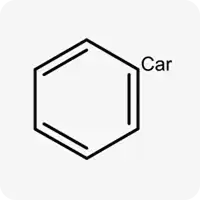 | Carbon atom in sp² hybridization, not part of a ring |
| Cs2 | 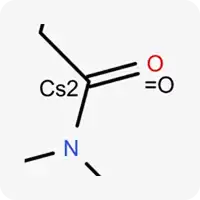 | Carbon atom in sp² hybridization, not part of a ring |
| Cs3 | 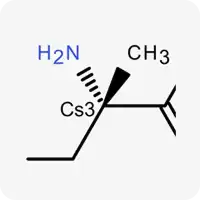 | Carbon atom in sp³ hybridization, not part of a ring |
| Csp | 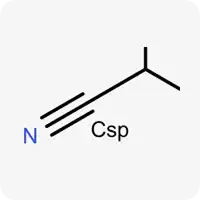 | Carbon atom in sp hybridization |
| Nac | 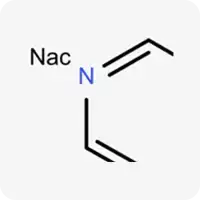 | Electrically neutral nitrogen, can accept hydrogen bonds |
| Nd+ | 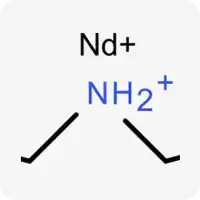 | Potentially ionizable nitrogen, contains a hydrogen atom and can act as a hydrogen bond donor |
| Nd0 | 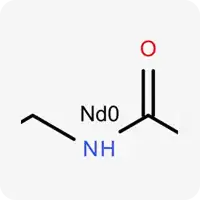 | Electrically neutral nitrogen, contains a hydrogen atom and can act as a hydrogen bond donor |
| O_a | 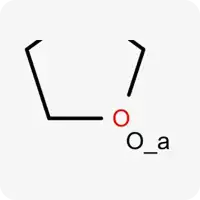 | Oxygen atom in sp³ hybridization, does not contain hydrogen atoms, acts as a hydrogen bond acceptor |
| SO2 | 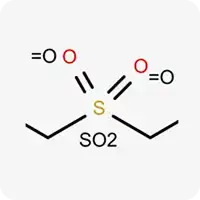 | Hexavalent sulfur atom |
| Sul | 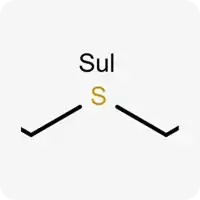 | Divalent sulfur atom |
4. Elements Limit
Restricts the number of specific elements allowed in the final scaffold:
- Nitrogen (N): 0–15, Default: 5
- Oxygen (O): 0–10, Default: 3
- Sulfur (S): 0–4, Default: 1
5. Maximum Number of N or O Atoms (0–30, Default: 8)
Limits the combined number of nitrogen and oxygen atoms.
6. Maximum Number of 3/4-Atom Rings (0–5, Default: 0)
Controls the presence of small rings (3-4-membered) in the scaffold.
7. Maximum Number of Conformations (1–5 or "All Positions", Default: 1)
- Defines the maximum number of unique conformations retained for a generated structure.
- Choosing “All Positions” allows storing all generated conformations, while lower values filter out less favorable ones.
8. Minimum Number of Heavy Atoms (5–20, Default: 7)
Defines minimum number of heavy atoms which should be present in generated structure to be shown in results.
9. Maximum Number of Positive Charges (0–4, Default: 0)
Controls the total number of atoms with a positive formal charge in the generated structure.
10. Maximum Number of Negative Charges (0–4, Default: 0)
Limits the total number of atoms with a negative formal charge in the generated structure.
11. Maximum Number of Charged Atoms (0–4, Default: 0)
Restricts the total number of positively or negatively charged atoms.
Advanced Parameters
Note: These settings should only be modified by experienced users.
1. Examples of Allowable Molecular Structures
Users can upload a set of molecular structures to disable undesirable medicinal chemistry filters (MCFs) automatically.
2. Approach Selection (MedChem/CatBoost, Default: MedChem)
- MedChem: Uses empirical weights for different terms in scoring function.
- CatBoost: Uses machine-learning-driven weights for different terms in scoring function.
3. Docking Score Limit (-12 to 0, Default: -5)
- Filters molecules based on predicted lg (Kd) values.
- More negative values (e.g., -12) indicate stronger affinity.
4. Output MedChem Filtration Mode (None/Simplified/Basic/Strict, Default: Strict)
Controls how strictly medicinal chemistry filters (MCFs) are applied:
- Strict – Recommended for drug-likeness validation, applies all MCF rules.
- Basic/Simplified – Loosens some restrictions to allow a broader chemical space.
- None – Disables all MCFs, allowing complete freedom in scaffold generation.
Best practice: Instead of fully disabling MCFs, upload allowable molecular structures and keep Strict Mode enabled to generate needed chemotypes without removing viable candidates.
5. Allow Minimization Skipping (Allow/Don't Allow, Default: Don't Allow)
- If enabled, both the initial and optimized positions of generated structures will be considered.
- This setting is useful when energy minimization significantly alters a scaffold's original position, potentially impacting its ability to form favorable interactions.
6. Minimum Local Difference (0.1–0.3 Å, Default: 0.2 Å)
Defines the minimum atomic displacement needed for two structures to be considered distinct.
7. Scoring Method (Expo/Sigmoid, Default: Expo)
Determines the way of balancing scoring function neural networks with each other:
- Expo – Uses an exponential decay function.
- Sigmoid – Applies a logistic function.
8. Number of Poses per Iteration (10–30, Default: 20)
- Sets how many intermediate structures are selected for next iteration.
- Higher values allow more structural diversity, but requires more computational time to perform complete calculations.
9. Maximum Acyclic Chain Length (0–6, Default: 4)
- Limits the length of molecular chains without rings within the generated scaffold.
- Ensures structures do not contain overly flexible fragments.
10. Maximum Clashes Number (5–20, Default: 12)
- Controls the number of steric clashes allowed in a generated molecule.
- A steric clash occurs when two non-bonded atoms are too close based on empirical distance thresholds.
- Lower values allow obtaining structures without steric hindrances but may lead to low structural diversity.
11. Maximum Number of Chiral Centers (0–3, Default: 0)
- Defines the maximum number of stereocenters in the output structure.
- Higher values allow more structural diversity, but synthetic feasibility may be affected.
Periphery Generation
Periphery Generation further modifies an existing scaffold by adding functional groups to increase molecular diversity and optimize interactions.
- Requires a predefined scaffold (either generated by Scaffold Generation or user-defined by Scaffold Placement).
- Expands the molecular chemical space while preserving defined scaffold structure.
Users can customize the process by adjusting basic and advanced parameters.
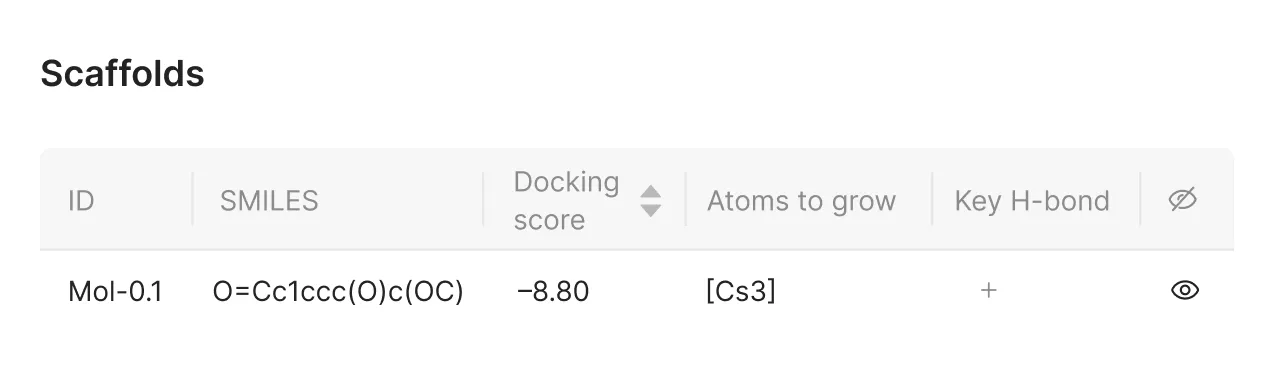
Atoms to Growth
In both Scaffold Generation and Periphery Generation, users need to define which atoms will serve as the starting points for molecular construction. This step allows generating only from selected growth points, decreasing number of obviously undesirable structures.
1. Opening Atom Selection
In the "Atoms to grow" column, click the "+" button to activate selection mode.
2. Visualizing Available Atoms
Once selection mode is active, the available atoms in the structure will be highlighted:
- Green – Selected as growth points for scaffold expansion or functional group attachment.
- Yellow – Available for selection but not chosen.

3. Modifying Atom Selection
- Click on an atom in the workspace window to change its status.
- Selected growth points define where the new molecular fragments or functional groups will be added.
4. Confirming Atom Selection
The chosen atoms appear in the corresponding column of the interface, allowing users to review and adjust their selections before proceeding.
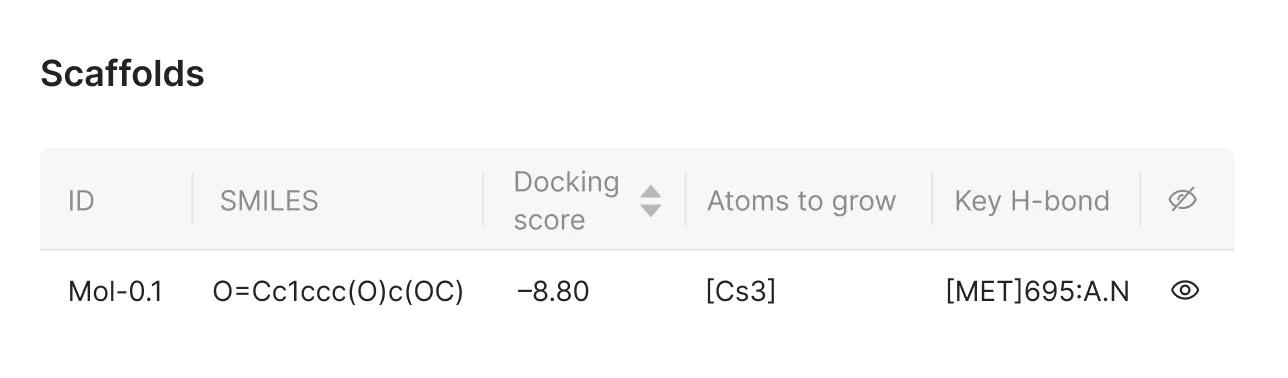
Key Hydrogen Bonds
In both Scaffold Generation and Periphery Generation, users can define key hydrogen bonds between the selected probe and the protein. This step ensures that the generated structure retains important hydrogen bond.
1. Opening Hydrogen Bond Selection
In the "Key H-bond" column, click the "+" button to activate interaction selection mode.
2. Visualizing Available Interactions
Once selection mode is active, hydrogen bonding sites will be highlighted:
- Green – Selected as a key hydrogen bonding site.
- Yellow – Available for selection but not chosen.

3. Modifying Interaction Selection
- Click on an interaction in the visualization window to change its status.
- Selected hydrogen bonding sites define which interaction should be retained during generation.
4. Confirming Interaction Selection
The chosen hydrogen bonding site will be listed in the corresponding column of the interface. Users can review and adjust their selections before proceeding.
Basic Parameters
1. Maximum Execution Time (1–168 h, Default: 96 h)
Limits the time allowed for functional group attachment.
2. Maximum Heavy Atoms to Add (1–30, Default: 20)
Defines the maximum number of non-hydrogen atoms that can be added during generation.
3. Types Limit
Controls the maximum number of each atom type allowed in the final structure.
| Atom Type | Example | Description |
|---|---|---|
| .=O | | Oxygen atom in sp² hybridization, acts as a hydrogen bond acceptor |
| C2r | | Carbon atom in sp² hybridization within a non-aromatic ring |
| C3r | | Carbon atom in sp³ hybridization within a ring |
| Car | | Carbon atom in sp² hybridization, not part of a ring |
| Cs2 | | Carbon atom in sp² hybridization, not part of a ring |
| Cs3 | | Carbon atom in sp³ hybridization, not part of a ring |
| Csp | | Carbon atom in sp hybridization |
| Nac | | Electrically neutral nitrogen, can accept hydrogen bonds |
| Nd+ | | Potentially ionizable nitrogen, contains a hydrogen atom and can act as a hydrogen bond donor |
| Nd0 | | Electrically neutral nitrogen, contains a hydrogen atom and can act as a hydrogen bond donor |
| O_a | | Oxygen atom in sp³ hybridization, does not contain hydrogen atoms, acts as a hydrogen bond acceptor |
| SO2 | | Hexavalent sulfur atom |
| Hal | 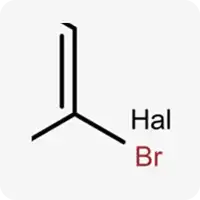 | Halogens (F, Cl, Br, I) |
| O_d | 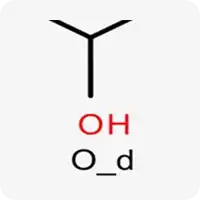 | Oxygen atom in sp³ hybridization, contains a hydrogen atom, acts as a hydrogen bond donor/acceptor |
4. Elements Limit
Restricts the number of specific elements allowed in the final structure:
- Nitrogen (N): 0–15, Default: 6
- Oxygen (O): 0–10, Default: 4
- Sulfur (S): 0–4, Default: 1
5. Maximum Number of N or O Atoms (0–30, Default: 10)
Limits the combined number of nitrogen and oxygen atoms.
6. Maximum Number of 3/4-Atom Rings (0–5, Default: 0)
Controls the presence of small rings (3-4-membered) in the structure.
7. Maximum Number of Conformations (1–5 or "All Positions", Default: 1)
- Defines the maximum number of unique conformations retained for a generated structure.
- Choosing “All Positions” allows storing all generated conformations, while lower values filter out less favorable ones.
8. Minimum Number of Heavy Atoms (5–20, Default: 10)
Ensures generated scaffolds have a minimum structural complexity.
9. Maximum Number of Positive Charges (0–4, Default: 0)
Controls the total number of atoms with a positive formal charge in the generated structure.
10. Maximum Number of Negative Charges (0–4, Default: 0)
Limits the total number of atoms with a negative formal charge in the generated structure.
11. Maximum Number of Charged Atoms (0–2, Default: 0)
Restricts the total number of positively or negatively charged atoms.
Advanced Parameters
Note: These settings should only be modified by experienced users.
1. Examples of Allowable Molecular Structures
Users can upload a set of molecular structures to disable undesirable medicinal chemistry filters (MCFs) automatically.
2. Approach Selection (MedChem/CatBoost, Default: MedChem)
- MedChem: Uses empirical weights for different terms in scoring function.
- CatBoost: Uses machine-learning-driven weights for different terms in scoring function.
3. Docking Score Limit (-12 to 0, Default: -5)
- Filters molecules based on predicted lg (Kd) values.
- More negative values (e.g., -12) indicate stronger affinity.
4. Output MedChem Filtration Mode (None/Simplified/Basic/Strict, Default: Strict)
Controls how strictly medicinal chemistry filters (MCFs) are applied: - Strict – Recommended for drug-likeness validation, applies all MCF rules. - Basic/Simplified – Loosens some restrictions to allow a broader chemical space. - None – Disables all MCFs, allowing complete freedom in structure generation.
Best practice: Instead of fully disabling MCFs, upload allowable molecular structures and keep Strict Mode enabled to generate needed chemotypes without removing viable candidates.
5. Allow Minimization Skipping (Allow/Don't Allow, Default: Don't Allow)
- If enabled, both the initial and optimized positions of generated structures will be considered.
- This setting is useful when energy minimization significantly alters a structure's original position, potentially impacting its ability to form favorable interactions.
6. Minimum Local Difference (0.1–0.3 Å, Default: 0.2 Å)
Defines the minimum atomic displacement needed for two structures to be considered distinct.
7. Scoring Method (Expo/Sigmoid, Default: Expo)
Determines the way of balancing scoring function neural networks with each other:
- Expo – Uses an exponential decay function.
- Sigmoid – Applies a logistic function.
8. Number of Poses per Iteration (10–30, Default: 20)
- Sets how many intermediate structures are selected for next iteration.
- Higher values allow more structural diversity, but requires more computational time to perform complete calculations.
9. Maximum Acyclic Chain Length (0–6, Default: 4)
- Limits the length of molecular chains without rings within the generated structure.
- Ensures structures do not contain overly flexible fragments.
10. Maximum Clashes Number (5–20, Default: 12)
- Controls the number of steric clashes allowed in a generated molecule.
- A steric clash occurs when two non-bonded atoms are too close based on empirical distance thresholds.
- Lower values allow obtaining structures without steric hindrances but may lead to low structural diversity.
11. Maximum Number of Chiral Centers (0–3, Default: 0)
- Defines the maximum number of stereocenters in the output structure.
- Higher values allow more structural diversity, but synthetic feasibility may be affected.